The Perils and Guardrails of Modifying Signalling Proteins in Bioassays
- Kiyan Afzali
- Mar 10
- 11 min read
Dr. Kiyan Afzali
‘What we observe is not nature itself, but nature exposed to our method of questioning.’
–Werner Heisenberg

1. Introduction
Life scientists dedicate enormous resources towards meticulously understanding and implementing the biological frameworks of the world for the ultimate benefit of living beings. This pursuit in the best cases has remarkably led to groundbreaking technological advancements, such as life-saving medical treatments. However, the process is often fraught with deficits in accuracy and efficiency, leaving considerable space for improvement. Drug discovery is an extremely costly, lengthy, and risky venture [1-4] that requires $1–2 billion [5-8] invested over 10–15 years [5, 9, 10] with only a 7% chance of approval for candidates entering clinical trials [11-13]. This low success rate is due to inadequacies primarily in efficacy and secondarily in safety [5, 11], highlighting the limited translational capacity of preclinical studies. A reproducibility crisis exists where 47% of researchers have failed to replicate a published study that was originally conducted by another team and 23% have failed to do so for a study of their own [14]. An important reason for this unfavourable situation is that laboratory experiments are performed under highly controlled, abstract, and specific conditions, while complex biological phenomena are profoundly dependent on the context of a multitude of interactions [15]. The most diverse and common molecular target of drug medicines is G protein-coupled receptors (GPCRs) [16], which are interrogated through bioassays as the pharmacologist’s ‘eyes to see’ [17]. As researchers of these prime targets, it is our duty to routinely clean our lens of misleading artefacts and focus our efforts towards the most physiologically relevant observations.
A pivotal concept for accurately interpreting the behaviour of GPCRs is their dependence on allostery. Allostery is a vital biochemical mechanism that has been described as ‘the second secret of life’, preceded only by the existence of the genome [18-20]. This process is a binding event that facilitates a change in the shape of a macromolecule which impacts a particular interaction at another region [21-24]. The behaviour occurs through a remodelling of the free energy landscape between thermodynamically coupled networks of amino acids from perturbations such as ligand binding, post-translational modification, or protein interactions, allowing proteins to sense and respond to changes in their environment [18, 20, 22, 23, 25]. GPCRs are considered nature’s ‘prototypical allosteric protein’ [21, 26, 27] because they are regulated in this fashion by ions, small molecules, lipids, receptor-activity modifying proteins, autoantibodies, membrane stretch, and even other GPCRs through dimerisation [28-30]. Rather than being static structures, GPCRs exist in an ensemble of different conformations that are interchangeable according to the free energy of the system [26, 27, 31]. Ligands binding to GPCRs can stabilise a distinct set of conformations, which promotes a certain pattern of interaction with various transducer proteins, leading to biased signalling [32].
Bioassays for interrogating GPCR structure and function typically rely on protein tags or modifications [33]. Tags are often peptides or entire proteins that are genetically engineered to be fused to a target protein to facilitate detection, purification, localisation, or characterisation of activity [34, 35]. Given the sensitive allosteric nature of GPCRs with their binding partners, tags or modifications are likely to influence their conformational dynamics and confound analyses of their properties. This notion has become more heavily supported by two recent studies that explored the ramifications of modifying the GPCR ternary complex on signalling [36, 37].
2. Receptor Modification Impacts Signalling
Direct modifications of GPCRs can impact their G protein coupling propensities. For the serotonin 5-HT2A receptor (5-HT2AR) with the Gαi/o family of transducers, Wright and colleagues [36] demonstrated that an N-terminal 3x hemagglutinin tag narrowed activation from all subtypes except Gαi1 to only Gαz (Figure 1). Although the tag was found to decrease 5-HT2AR density, this impact on G protein coupling was primarily attributed to differences in the conformational dynamics of the receptor rather than a lowered sensitivity for detecting weaker interactions, as increasing receptor transfection only enhanced activation of GαoB and Gαz. These findings suggest that this tag, and potentially others, could have impacted the results of previous studies determining the 5-HT2AR structure, its molecular interactions with various ligands and canonical Gαq transducer, and conformational changes involved in active-state transitions [38]. This limited detection and modelling of 5-HT2AR-Gαi/o coupling could mistakenly divert attention from a potentially important pathway that has already shown relevance in the pathophysiology of schizophrenia and mechanism of psychedelic drugs [39-41]. The authors recommend that results obtained using a tagged receptor should be confirmed with an untagged version, and that whether identified coupling signatures have physiological relevance be further assessed in more appropriate biological systems [36].
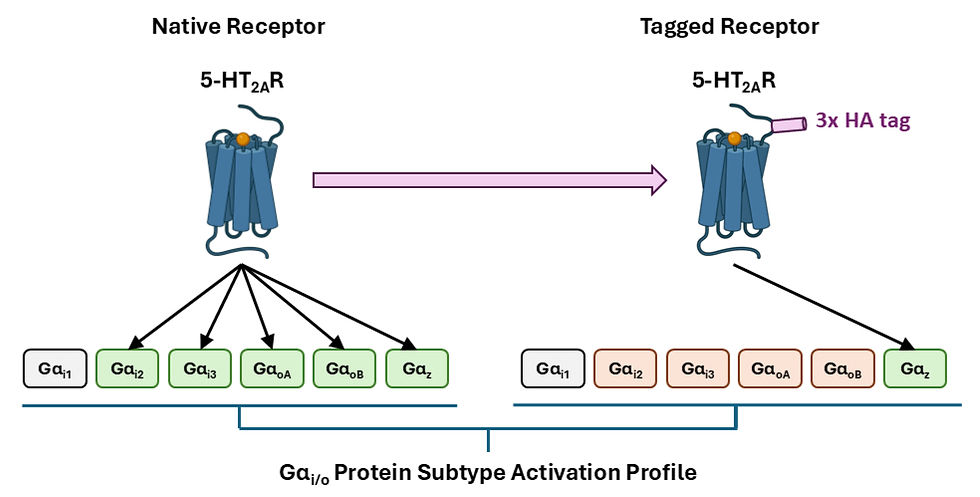
Figure 1. Modifying the serotonin 5-HT2A receptor (5-HT2AR) with a 3x hemagglutinin (HA) tag as used in structural studies narrowed the activation of Gαi/o protein subtypes from all except Gαi1 to only Gαz in a recent study by Wright and colleagues [36].
3. G Protein Modification Influences Signalling
Similar to GPCRs, modification of G proteins can influence transducer coupling signatures. A standard format of GPCR biosensors incorporates a luminescent tag on the G protein Gα subunit and Gβγ dimer, such that GPCR activation results in dissociation of these proteins and a decrease in signal [42, 43]. Wright and colleagues [36] showed that these heterotrimer-based biosensors for the 5-HT7A/B receptors (5-HT7A/BRs) were only able to detect interaction with the strongest coupling partner Gαs (Figure 2A), and for the glucagon-like-peptide-1 receptor (GLP-1R) even reported unproductive interactions involving conformational rearrangements of the Gαβγ protein without signalling. In contrast, biosensors of a strategically different format measuring unmodified active Gα subunits through G protein effector translocation to the plasma membrane captured additional weaker couplings with Gαi/o/z and Gαq/11/14/15 for the 5-HT7A/BRs (Figure 2A), and was immune to such irrelevant signals for the GLP-1R. Moreover, these alternate biosensors reported different Gαi/o coupling rankings between four of six subtypes for the 5-HT2AR, with the exceptions being the strongest and weakest interactions to Gαz and Gαi1, respectively (Figure 2B), as well as different coupling preferences between specific Gβγ pairs for the muscarinic acetylcholine M3 receptor (M3R) (Figure 2C). These findings suggest that GPCR biosensors consisting of modified G proteins, which have been implemented in pharmacological research and development for decades, may not be suitable for detecting and accurately evaluating the complete range of coupling interactions. The authors emphasise the importance of understanding the advantages and limitations of different assay technologies and taking these factors into consideration when interpreting results and drawing conclusions from studies [36].
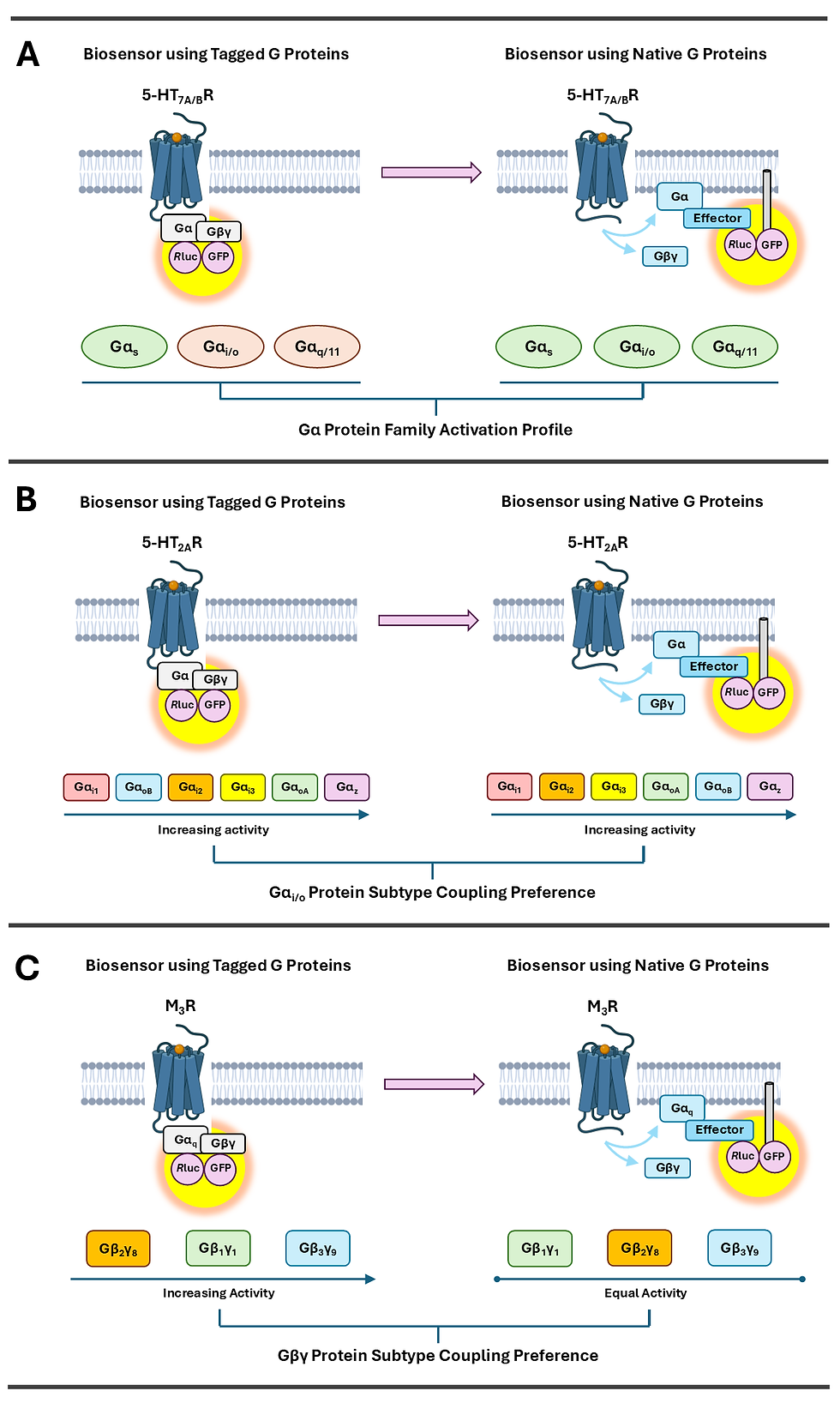
Figure 2. Modifying G proteins with luminescent tags as used in biosensors altered the G protein coupling profiles of the (A) serotonin 5-HT7A/B receptors (5-HT7A/BRs) (subtypes activated are coloured in green and otherwise red), (B) serotonin 5-HT2A receptor (5-HT2AR), and (C) muscarinic acetylcholine M3 receptor (M3R) in a recent study by Wright and colleagues [36]. Rluc: Renilla luciferase, GFP: Green Fluorescent Protein.
Another format of biosensors involves the use of mini-G proteins, which are genetically engineered portions of Gα subunits designed to stabilise GPCRs in their active conformations [44-46]. Unlike native Gα subunits, mini-G proteins do not bind Gβγ dimers or participate in nucleotide exchange [45, 46]. Manchanda and colleagues [37] showed that for the glucagon-like-peptide-1 receptor (GLP-1R), mini-Gs blocked β-arrestin recruitment, receptor internalisation, and endosomal trafficking, while other mini-G protein subtypes produced weaker or no such inhibition in correlation with their coupling selectivity. Similar effects of varying intensity were observed for several other GPCRs, including the β2-adrenergic receptor (β2-AR), μ-opioid receptor (MOR), free fatty acid receptor 2 (FFA2R), and glucose-dependent insulinotropic polypeptide receptor (GIPR) (Figure 3). Conversely, internalisation in the presence of mini-G proteins was maintained for a non-GPCR, the epidermal growth factor receptor (EGFR), which is instead a receptor tyrosine kinase. In contrast with mini-G proteins, the use of a nanobody, Nb37, previously developed to bind Gαs in complex with active GPCRs, enabled detection of GLP-1R activation both from the plasma membrane and endosomes. These findings suggest that the use of mini-G proteins could have hindered the accurate quantification of GPCR signalling from subcellular compartments in previous studies [44, 47-49]. Additionally, as mini-G proteins are increasingly implemented in structural determinations of GPCRs and fused with fluorescent or luminescent tags for evaluating signalling particular to various G protein subtypes, it is possible that they could influence the results of these studies with unexpected effects as well [50-52]. To circumvent their identified problem, the authors recommend checking GPCR internalisation rates to determine tolerable expression levels of mini-G proteins and validating results with other tools, such as those involving full-length G protein constructs [37].
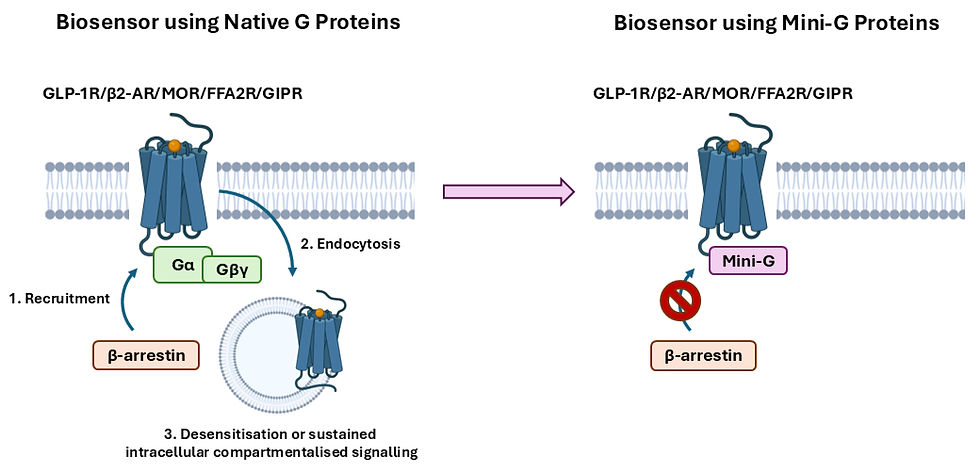
Figure 3. Mini-G proteins as modified versions of Gα proteins inhibited β-arrestin recruitment, receptor internalisation, and endosomal trafficking for the glucagon-like-peptide-1 receptor (GLP-1R), β2-adrenergic receptor (β2-AR), µ-opioid receptor (MOR), free fatty acid receptor 2 (FFA2R), and glucose-dependent insulinotropic polypeptide receptor (GIPR) in a recent study by Manchanda and colleagues [37].
3. Conclusion and Outlook
Overall, these latest studies demonstrate that modifying GPCRs or their binding partners for bioassays can alter their interactions and produce misleading results. This conundrum for the pharmacologist is analogous to the physicist’s [53] or social scientist’s [54-56] observer’s paradox, where determinations of quantum mechanical systems or human behaviour, respectively, are influenced by the act of observation [57]. Fortunately, GPCR researchers have solutions at their disposal to overcome this challenge, essentially in which, the allosteric nature of GPCRs must be respected. Moving forward, implementing and developing assay technologies that strategically operate using unmodified versions of these proteins and their binding partners will be important for ensuring that findings accurately reflect native biological processes and preclinical research can more reliably translate into therapeutic success.
References
1. Freeman BB, 3rd, Yang L, Rankovic Z. Practical approaches to evaluating and optimizing brain exposure in early drug discovery. Eur J Med Chem. 2019;182:111643.
2. Wager TT, Hou X, Verhoest PR, Villalobos A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem Neurosci. 2010;1(6):435-49.
3. Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3(8):711-5.
4. Butlen-Ducuing F, Pétavy F, Guizzaro L, Zienowicz M, Salmonson T, Haas M, et al. Challenges in drug development for central nervous system disorders: a European Medicines Agency perspective. Nature Reviews Drug Discovery. 2016;15(12):813-814.
5. Sun D, Gao W, Hu H, Zhou S. Why 90% of clinical drug development fails and how to improve it? Acta Pharmaceutica Sinica B. 2022;12(7):3049-3062.
6. Wouters OJ, McKee M, Luyten J. Estimated Research and Development Investment Needed to Bring a New Medicine to Market, 2009-2018. Jama. 2020;323(9):844-853.
7. DiMasi JA, Grabowski HG, Hansen RW. Innovation in the pharmaceutical industry: New estimates of R&D costs. J Health Econ. 2016;47:20-33.
8. Schlander M, Hernandez-Villafuerte K, Cheng CY, Mestre-Ferrandiz J, Baumann M. How Much Does It Cost to Research and Develop a New Drug? A Systematic Review and Assessment. Pharmacoeconomics. 2021;39(11):1243-1269.
9. Brown DG, Wobst HJ, Kapoor A, Kenna LA, Southall N. Clinical development times for innovative drugs. Nat Rev Drug Discov. 2022;21(11):793-794.
10. Mohs RC, Greig NH. Drug discovery and development: Role of basic biological research. Alzheimers Dement (N Y). 2017;3(4):651-657.
11. Dowden H, Munro J. Trends in clinical success rates and therapeutic focus. Nat Rev Drug Discov. 2019;18(7):495-496.
12. Wong CH, Siah KW, Lo AW. Estimation of clinical trial success rates and related parameters. Biostatistics. 2019;20(2):273-286.
13. Yamaguchi S, Kaneko M, Narukawa M. Approval success rates of drug candidates based on target, action, modality, application, and their combinations. Clin Transl Sci. 2021;14(3):1113-1122.
14. Cobey KD, Ebrahimzadeh S, Page MJ, Thibault RT, Nguyen PY, Abu-Dalfa F, et al. Biomedical researchers' perspectives on the reproducibility of research. PLoS Biol. 2024;22(11):e3002870.
15. Hunter P. The reproducibility "crisis": Reaction to replication crisis should not stifle innovation. EMBO Rep. 2017;18(9):1493-1496.
16. Liu S, Anderson PJ, Rajagopal S, Lefkowitz RJ, Rockman HA. G Protein-Coupled Receptors: A Century of Research and Discovery. Circ Res. 2024;135(1):174-197.
17. Kenakin TP. Chapter 2 - How different tissues process drug response. In: Kenakin TP, editor A Pharmacology Primer (Sixth Edition). Academic Press; 2022. p. 23-45.
18. Lu S, He X, Ni D, Zhang J. Allosteric Modulator Discovery: From Serendipity to Structure-Based Design. Journal of Medicinal Chemistry. 2019;62(14):6405-6421.
19. Fenton AW. Allostery: an illustrated definition for the 'second secret of life'. Trends Biochem Sci. 2008;33(9):420-5.
20. Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, Christopoulos A, et al. Allosteric modulation of seven transmembrane spanning receptors: theory, practice, and opportunities for central nervous system drug discovery. J Med Chem. 2012;55(4):1445-64.
21. Persechino M, Hedderich JB, Kolb P, Hilger D. Allosteric modulation of GPCRs: From structural insights to in silico drug discovery. Pharmacol Ther. 2022;237:108242.
22. Chatzigoulas A, Cournia Z. Rational design of allosteric modulators: Challenges and successes. WIREs Computational Molecular Science. 2021;11(6):e1529.
23. Wodak SJ, Paci E, Dokholyan NV, Berezovsky IN, Horovitz A, Li J, et al. Allostery in Its Many Disguises: From Theory to Applications. Structure. 2019;27(4):566-578.
24. Changeux J-P, Christopoulos A. Allosteric Modulation as a Unifying Mechanism for Receptor Function and Regulation. Cell. 2016;166(5):1084-1102.
25. Motlagh HN, Wrabl JO, Li J, Hilser VJ. The ensemble nature of allostery. Nature. 2014;508(7496):331-339.
26. Kenakin TP. Seven transmembrane receptors as nature's prototype allosteric protein: de-emphasizing the geography of binding. Mol Pharmacol. 2008;74(3):541-3.
27. Kenakin TP. Biased signalling and allosteric machines: new vistas and challenges for drug discovery. Br J Pharmacol. 2012;165(6):1659-1669.
28. Shen S, Zhao C, Wu C, Sun S, Li Z, Yan W, et al. Allosteric modulation of G protein-coupled receptor signaling. Frontiers in Endocrinology. 2023;14.
29. Slosky LM, Caron MG, Barak LS. Biased Allosteric Modulators: New Frontiers in GPCR Drug Discovery. Trends in Pharmacological Sciences.
30. van der Westhuizen ET, Valant C, Sexton PM, Christopoulos A. Endogenous allosteric modulators of G protein-coupled receptors. J Pharmacol Exp Ther. 2015;353(2):246-60.
31. Kenakin T. The mass action equation in pharmacology. Br J Clin Pharmacol. 2016;81(1):41-51.
32. Kenakin T. Biased Receptor Signaling in Drug Discovery. Pharmacol Rev. 2019;71(2):267-315.
33. Pearce A, Redfern-Nichols T, Wills E, Rosa M, Manulak I, Sisk C, et al. Quantitative approaches for studying G protein-coupled receptor signalling and pharmacology. J Cell Sci. 2025;138(1).
34. Kosobokova EN, Skrypnik KA, Kosorukov VS. Overview of Fusion Tags for Recombinant Proteins. Biochemistry (Mosc). 2016;81(3):187-200.
35. Dave K, Gelman H, Thu CT, Guin D, Gruebele M. The Effect of Fluorescent Protein Tags on Phosphoglycerate Kinase Stability Is Nonadditive. J Phys Chem B. 2016;120(11):2878-85.
36. Wright SC, Avet C, Gaitonde SA, Muneta-Arrate I, Le Gouill C, Hogue M, et al. Conformation- and activation-based BRET sensors differentially report on GPCR-G protein coupling. Science signaling. 2024;17(841):eadi4747.
37. Manchanda Y, ElEid L, Oqua AI, Ramchunder Z, Choi J, Shchepinova MM, et al. Engineered mini-G proteins block the internalization of cognate GPCRs and disrupt downstream intracellular signaling. Science signaling. 2024;17(843):eabq7038.
38. Kim K, Che T, Panova O, DiBerto JF, Lyu J, Krumm BE, et al. Structure of a Hallucinogen-Activated Gq-Coupled 5-HT2A Serotonin Receptor. Cell. 2020;182(6):1574-1588.e19.
39. García-Bea A, Miranda-Azpiazu P, Muguruza C, Marmolejo-Martinez-Artesero S, Diez-Alarcia R, Gabilondo AM, et al. Serotonin 5-HT2A receptor expression and functionality in postmortem frontal cortex of subjects with schizophrenia: Selective biased agonism via Gαi1-proteins. Eur Neuropsychopharmacol. 2019;29(12):1453-1463.
40. González-Maeso J, Weisstaub NV, Zhou M, Chan P, Ivic L, Ang R, et al. Hallucinogens recruit specific cortical 5-HT2A receptor-mediated signaling pathways to affect behavior. Neuron. 2007;53(3):439-52.
41. Muneta-Arrate I, Diez-Alarcia R, Horrillo I, Meana JJ. Pimavanserin exhibits serotonin 5-HT2A receptor inverse agonism for Gαi1- and neutral antagonism for Gαq/11-proteins in human brain cortex. Eur Neuropsychopharmacol. 2020;36:83-89.
42. Olsen RHJ, English JG. Advancements in G protein-coupled receptor biosensors to study GPCR-G protein coupling. Br J Pharmacol. 2023;180(11):1433-1443.
43. Olsen RHJ, DiBerto JF, English JG, Glaudin AM, Krumm BE, Slocum ST, et al. TRUPATH, an open-source biosensor platform for interrogating the GPCR transducerome. Nature chemical biology. 2020;16(8):841-849.
44. Wan Q, Okashah N, Inoue A, Nehmé R, Carpenter B, Tate CG, et al. Mini G protein probes for active G protein-coupled receptors (GPCRs) in live cells. J Biol Chem. 2018;293(19):7466-7473.
45. Nehmé R, Carpenter B, Singhal A, Strege A, Edwards PC, White CF, et al. Mini-G proteins: Novel tools for studying GPCRs in their active conformation. PLoS One. 2017;12(4):e0175642.
46. Carpenter B, Tate CG. Engineering a minimal G protein to facilitate crystallisation of G protein-coupled receptors in their active conformation. Protein Eng Des Sel. 2016;29(12):583-594.
47. Crilly SE, Ko W, Weinberg ZY,Puthenveedu MA. Conformational specificity of opioid receptors is determined by subcellular location irrespective of agonist. Elife. 2021;10.
48. Truong ME, Bilekova S, Choksi SP, Li W, Bugaj LJ, Xu K, et al. Vertebrate cells differentially interpret ciliary and extraciliary cAMP. Cell. 2021;184(11):2911-2926.e18.
49. Jimenez-Vargas NN, Gong J, Wisdom MJ, Jensen DD, Latorre R, Hegron A, et al. Endosomal signaling of delta opioid receptors is an endogenous mechanism and therapeutic target for relief from inflammatory pain. Proc Natl Acad Sci U S A. 2020;117(26):15281-15292.
50. García-Nafría J, Tate CG. Cryo-EM structures of GPCRs coupled to Gs, Gi and Go. Mol Cell Endocrinol. 2019;488:1-13.
51. Rößler P, Mayer D, Tsai CJ, Veprintsev DB, Schertler GFX, Gossert AD. GPCR Activation States Induced by Nanobodies and Mini-G Proteins Compared by NMR Spectroscopy. Molecules. 2020;25(24).
52. Teng X, Chen S, Wang Q, Chen Z, Wang X, Huang N, et al. Structural insights into G protein activation by D1 dopamine receptor. Science advances. 2022;8(23):eabo4158.
53. Carroll S. Why even physicists still don't understand quantum theory 100 years on. Nature. 2025;638(8049):31-34.
54. Franzen MM, Alder ML, Dreyer F, Köpp W, Buchholz MB. Being right vs. getting it right: orientation to being recorded in psychotherapeutic interaction as disaffiliative vs. affiliative practice. Front Psychol. 2023;14:1254555.
55. Hazel S. The paradox from within: research participants doing-being-observed. Qualitative Research. 2015;16(4):446-467.
56. Wilner W. Participatory experience: the participant observer paradox. Am J Psychoanal. 1987;47(4):342-57.
57. Lowe D. Fluorescent Tags Are Basically Never Silent. In the Pipeline (Science, 2024).
Acknowledgements
Figures in this article were created with the assistance of BioRender.com
Comments